This article is based on research I did as a freshman undergrad, see the full project here.
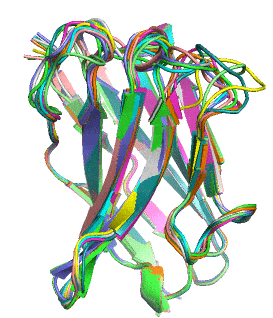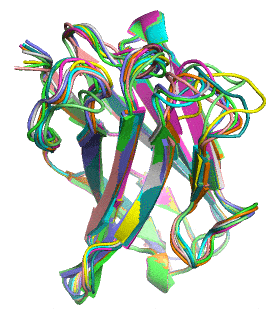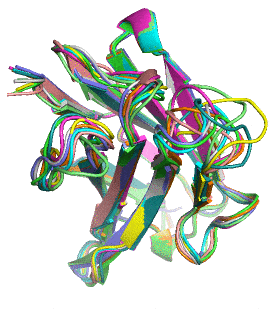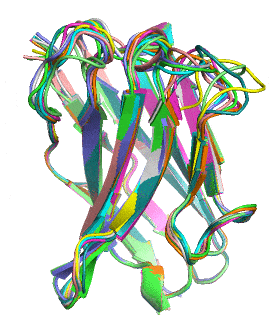
Proteins naturally fold into their lowest energy configuration, just like a ball rolling down a hill to a lower potential. This folded structure gives proteins their function, similar to how which lock a key can open is determined by its shape, not the metal that makes it up. Theoretically it is possible to calculate how a protein will fold just by using simple electrostatic attraction of all a protein’s atoms to each other. However, modeling this process on an atom-by-atom basis is NP-hard, meaning that it cannot be easily scaled to average size proteins without a supercomputer.
Rosetta is a software suite for modeling and simulating complex proteins which is designed to help solve such problems. Rosetta reduces the computational complexity of this problem by dividing the sequence into fragments and substituting fragments of common folding patterns into the protein. It simulates random conformations in space and performs a Monte Carlo algorithm to find the lowest energy conformation. Essentially, random conformations are simulated to reduced the free energy in an iterative process until optimal conformations are obtained. If the change reduced the free energy, Rosetta continues from the new conformation. If the change increased the free energy, the simulation reverts to the old conformation.
Homology modeling can be used to predict the structure of a protein from just its amino acid sequence very quickly and accurately. Rosetta has builtin homology modeling support through rosettacm. Homology modeling uses information from other similar protein sequences structures to see how differences from the known structure will effect the new structure of the protein you want. Because you use previously mapped protein structures the quality and accurately of the models you produce will depend on how similar the structures you find are to your protein of interest. More similar proteins with less addition gaps lead to faster and more accurate results.
The first step is to make a fasta file for your target sequence. This is simple
and can be done manually. An example is shown below. Change name to the name
of your protein this should match the name of your file (eg 5vnv.fasta).
>name
EVQLQASGGGFVQPGGSLRLSCAASGSTSRQYDMGWFRQAPGKEREFVSAISSNQDQPPYYADSVKGRFTISRDNSKNTVYLQMNSLRAEDTATYYCAFKQHHANGAYWGQGTQVTVSS
Next you want to get the PDB files for around 2-5 similar proteins. This can be done using a website called blast. Simply enter the target proteins sequence, select to only show proteins with structures already mapped, and press enter. This will query their database of known proteins and find the ones most similar. You want to select proteins which are most similar and have the fewest gaps. When choosing proteins it is often best to use the ones with the least amount of addition gaps. Rosetta has a harder time adding new amino acids compared to deleting ones or swapping residues out. So it is commonly a good idea to select sequences which have a lower similarity score but have less gaps and make less additions. Depending on the protein you are trying to simulate there might be more options which are very similar so less than 5 proteins would be needed.
Once you have selected your similar proteins you need to use another website
called the protein databank to download the PDB files you want. However this
PDB from the protein databank most likely contains chains you do not care
about, so you have to remove them for rosetta to understand which chains you
want to simulate. Luckily rosetta comes with a python script called
clean_pdb.py to do exactly this. Simply give this script the PDB file name
and the chain you want to keep. This program will also produce a fasta file for
the sequence of the chain which is needed in the next step.
Yet another website, clustal omega, is used to combine all the similar and the target protein sequences so they can be easily compared by rosetta. The file it outputs however can not be understood by rosetta, so I wrote a python script to convert this clustal file into one rosetta can read, grishin.
This grishin alignment file and the similar protein PDBs are then given to rosetta to generate a protein threaded PDBs for each input PDB. These threaded PDBs, the target sequence fasta file and a rosettacm XML file are then all given to another rosetta program to generate the target protein’s PDB file. It is recommended you generate anywhere from 50-200 PDBs depending on how quickly the results seem to converge.
$ rosetta/main/source/bin/partial_thread.default.linuxgccrelease -in:file:fasta molxa3.fasta -in:file:alignment molxa3_6dbeA.grishin -in:file:template_pdb 6dbeA.pdb
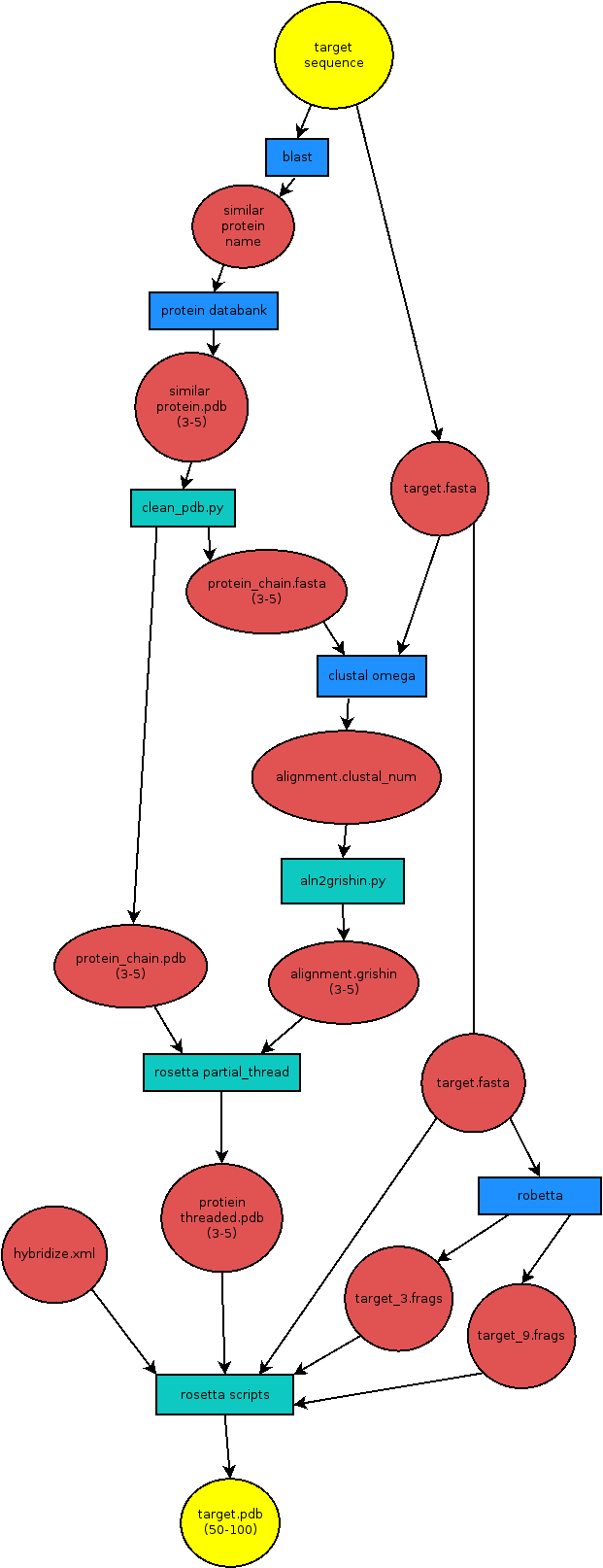
A full flow-diagram for this process can be seen below: